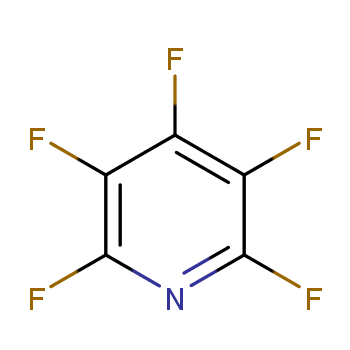
Pentafluoropyridine, also known as perfluoropyridine, has the chemical name Pentafluoropyridine, CAS: 700-16-3, and the molecular formula C5F5N. It features a six-membered aromatic ring where five fluorine atoms replace all hydrogen atoms on the carbon atoms. As a significant perfluoroaromatic compound, the introduction of fluorine atoms and heteroatoms into its molecule markedly alters its structure and electronic distribution. This change imparts unique physicochemical properties to pentafluoropyridine and affects its excitation mechanism and relaxation processes. The introduction of fluorine atoms or radicals increases the atomic nucleus mass, thus lowering the excitation energy, leading to a notable red shift in the absorption spectrum compared to benzene. Additionally, fluorine atoms enhance the π-bonding and antibonding characteristics in aromatic rings or heterocycles. With increasing fluorine atoms, the C—F bond length gradually shortens, making the molecular structure of fluorinated aromatic compounds more stable and aromatic.
The synthesis of pentafluoropyridine was first reported in the early 1960s, involving the defluorination of perfluoropiperidine. In this process, perfluoropiperidine was prepared by reacting pyridine with anhydrous hydrogen fluoride in an electrochemical synthesis at high temperatures with metals like iron or nickel. Subsequently, pentafluoropyridine was obtained by chromatographic separation. When iron was used as a reducing agent, the yield of pentafluoropyridine was 26%, whereas with nickel, the yield was slightly lower at 12%.

In 1964 and 1965, Chambers et al. and Banks et al. respectively published similar synthesis methods for pentafluoropyridine. The authors heated pentachloropyridine and anhydrous potassium fluoride in a high-pressure vessel to produce pentafluoropyridine. Pentachloropyridine was prepared by reacting pyridine with phosphorus pentachloride. This method produced a mixture of products, which could be separated by distillation.

The total amount of halogenated products was about 90%, and the product ratio could be adjusted by changing the temperature and reaction time. Under optimal conditions, the yield of pentafluoropyridine was 83%. This method remains the gold standard for commercial synthesis of pentafluoropyridine.
Since the mid-1960s, there has been little progress in alternative methods for synthesizing pentafluoropyridine. About 20 years later, Banks et al. pyrolyzed pentafluoro(trichloromethyl)benzene and 4-dichloraminotetrafluoropyridine under nitrogen protection to obtain trace amounts of pentafluoropyridine. In the same year, Coe and Sleigh pyrolyzed various pyrrolidines in the presence of iron. This reaction produced a mixture of products, similar to Banks' report, with very low yields of pentafluoropyridine (<12%). Later in 1982, Plevey and colleagues achieved a 40% yield of pentafluoropyridine by fluorinating pyridine with cesium tetrafluorocobaltate (III). However, they also encountered challenges, as yields decreased when the reaction scale exceeded 5 grams.

In 2004, Bardin et al. published a final example where they obtained pentafluoropyridine by dehalogenating C5Cl4F5N in the presence of iron or zinc. In all cases, a mixture of products was obtained, and the non-separated yield was determined by GLC analysis, as shown in the table below.

Using 2,4,6-trifluoro-3,5-dichloropyridine as the raw material and KF as the fluorinating agent, Zhong Xuhui et al. focused on the synthesis of pentafluoropyridine. Through research on the fluorination reaction catalyst, they screened new catalysts to lower the reaction temperature, laying the foundation for the industrialization of such challenging fluorination reactions. By exploring the experimental mechanism, they identified a new combination catalyst, tetraphenyl phosphonium bromide and anhydrous AlCl3 in a 3:1 mass ratio, applied to the reaction with 2,4,6-trifluoro-3,5-dichloropyridine and KF as the fluorinating agent, achieving favorable experimental results. Optimization of the experimental parameters yielded: KF: trifluoro-dichloropyridine: tetraphenyl phosphonium bromide: AlCl3 = 30g: 150g: 3g: 1g, reaction temperature of 240°C, reaction time of 20 hours, with 3-chloro-tetrafluoropyridine at 15%, pentafluoropyridine at 10%, and catalyst selectivity at 50%.
The chemical transformations of pentafluoropyridine have been well documented, including notable methods such as CF bond activation, photo-oxidation-reduction reactions, dehydrofluorination, and nucleophilic addition. Due to the variety of nucleophilic reagents, the known chemical transformations are highly flexible. Additionally, pentafluoropyridine exhibits unique regioselectivity, with metathetical additions at the 4-position being distinctive under mild basic conditions, and nucleophilic reagents of types O-, N-, S-, and C-. 2,6-Ordered additions can also be accomplished, while 3,5-Disubstitution occurs separately.

Research by Vlasov et al. shows that pentafluoropyridine can undergo reversible substitution reactions with fluorides (F?) under basic conditions, facilitated by good nucleophiles such as phenol. Further studies using DFT modeling explored the reversible substitution effects of electron-rich and electron-poor aromatic ethers at the 2,6-positions of pentafluoropyridine. Brittain et al. utilized these findings, employing pentafluoropyridine as an effective protecting group for various phenolic compounds, which could undergo cleavage or deprotection reactions under mild conditions.

Pentafluoropyridine has been used as a nucleophilic fluorinating reagent for alkyl halides (from right to left in the figure) and for deoxygenation of carboxylic acids (from left to right in the figure) to prepare multifunctional substrate pools. The addition of pentafluoropyridine and N, N-dimethylaminopyridine generates air-stable fluorination salts, enabling the fluorination of organic halides (R-X). Additionally, nucleophilic addition of pentafluoropyridine with carboxylic acids readily generates acyl fluorides in situ. The ester intermediate then cleaves from the non-chelated fluoride, yielding phenol as a by-product and the desired acyl fluoride. This strategy extends to using pentafluoropyridine as a coupling agent in one-pot synthesis of amides and esters.

Purposeful manipulation of pentafluoropyridine includes site-selective dehalogenation and halogen exchange (halex), as illustrated. These processes yield regioselective substitutions depending on reaction conditions. Senaweera et al. utilized a flow reactor design to prepare dehydrofluorinated pentafluoropyridine, achieving high photocatalytic turnover under low Ir complex loading, demonstrating the scalability of such useful intermediates. Halex reverse substitution at the 4-position of pentafluoropyridine with LiCl was achieved, with additional exchange with chlorine controlled by kinetics and thermodynamics.

[4] https://baike.baidu.com/item/%E4%BA%94%E6%B0%9F%E5%90%A1%E5%95%B6
|
 |